

Descrits defectes genètics en un enzim associat a la biosíntesi del coenzim A
En un estudi on participen investigadors del grup de Biologia Molecular de Llevats de l'Institut de Biotecnologia i Biomedicina (UAB) s'ha descrit per primera vegada un error congènit ultrarar en el metabolisme del coenzim A, involucrat en els processos d'obtenció d'energia en l'organisme. Aquesta recerca crea els fonaments per a detectar precoçment l'error i poder esmenar-lo abans que causi patologia.
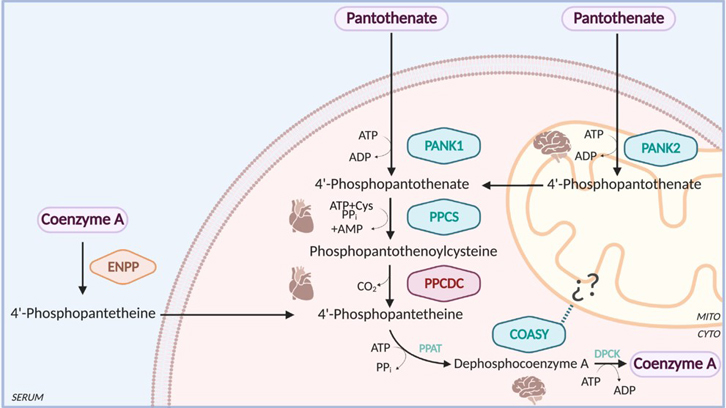
El coenzim A (CoA) és un cofactor essencial involucrat en una gran varietat de processos metabòlics essencials per a l'obtenció d'energia. Tant eucariotes com procariotes sintetitzen CoA a partir del pantotenat, conegut també com a vitamina B5, mitjançant una via metabòlica gairebé universal. En humans, aquesta via implica cinc passos enzimàtics que involucren quatre enzims: pantotenat quinasa (PANK), 4'-fosfopantotenoilcisteïna sintetasa (PPCS), fosfopantotenoilcisteïna descarboxilasa (PPCDC) i CoA sintasa (COASY). Fins ara es coneixien defectes genètics en 3 dels 4 enzims esmentats, sent l'excepció la PPCDC. En general aquests defectes donen lloc a una patologia de caràcter sever. La PPCDC codifica una cisteïna descarboxilasa que utilitza flavin mononucleòtid (FMN) com a cofactor i catalitza la descarboxilació de 4'-fosfopantotenoilcisteïna a 4'-fosfopanteteïna. En els humans, la forma nativa és un homotrímer de 204 aminoàcids en què el centre actiu es forma a la interfície de cada dues subunitats.
El primer cas descrit de defectes genètics associats a la PPCDC va ser publicat recentment a la revista Journal of Inherited Metabolic Disease com a resultat d'una col·laboració multicèntrica entre diversos laboratoris de Madrid i València que ha comptat a més amb la participació dels investigadors de la UAB. Es tracta d'un cas detectat en dues germanes, que van presentar severs problemes cardíacs i van morir molt prematurament, que es va poder associar a variants bialèliques (p.Thr53Pro i p.Ala95Val) de la PPCDC. Aquestes variants afecten residus altament conservats a través de diferents espècies; p.Thr53Pro està involucrat en la unió del mononucleòtid de flavina, i p.Ala95Val és probablement una mutació desestabilitzadora. Com a resultat, els fibroblasts derivats d'aquests pacients van mostrar una absència de proteïna PPCDC i una reducció de gairebé el 50% als nivells de CoA. A més, les cèl·lules presentaven clars problemes de carència energètica acompanyats de defectes en la respiració mitocondrial.
Aquests resultats van ser recolzats pels experiments realitzats emprant un model de llevats deficitari en PPCDC i, per tant, en la biosíntesi de CoA, posat a punt pel grup de Biologia Molecular de Llevats de l'Institut de Biotecnologia i Biomedicina (UAB) i dirigit pel Dr. Joaquín Ariño. Es tracta, doncs, d'un treball que descriu per primera vegada un nou i ultrarar error congènit del metabolisme, de greus conseqüències, causat per variants patogèniques de la PPCDC. D'aquesta manera, s'asseuen les bases per a una possible detecció precoç que permetria una teràpia de suplementació que eludís la deficiència en PPCDC.
Aquest treball posa també de manifest la importància dels estudis col·laboratius multidisciplinaris en l'elucidació de les bases genètiques de les malalties ultrarares i emfatitza la importància del desenvolupament de models de recerca en eucariotes unicel·lulars, com els llevats, l'aplicació dels quals resulta clau en la investigació de sistemes més complexos.
Departament de Bioquímica i Biologia Molecular, Universitat Autònoma de Barcelona.
Institut de Biotecnologia i Biomedicina (IBB-UAB), Universitat Autònoma de Barcelona.
Referències
Bravo-Alonso I, Morin M, Arribas-Carreira L, Álvarez M, Pedrón-Giner C, Soletto L, Santolaria C, Ramón-Maiques S, Ugarte M, Rodríguez-Pombo P, Ariño J, Moreno-Pelayo MÁ, Pérez B. Pathogenic variants of the coenzyme A biosynthesis-associated enzyme phosphopantothenoylcysteine decarboxylase cause autosomal-recessive dilated cardiomyopathy. J Inherit Metab Dis. 2022 Dec 23. doi: 10.1002/jimd.12584. Epub ahead of print. PMID: 36564894.
