

Genetic defects described for the first time in an enzyme associated with coenzyme A biosynthesis
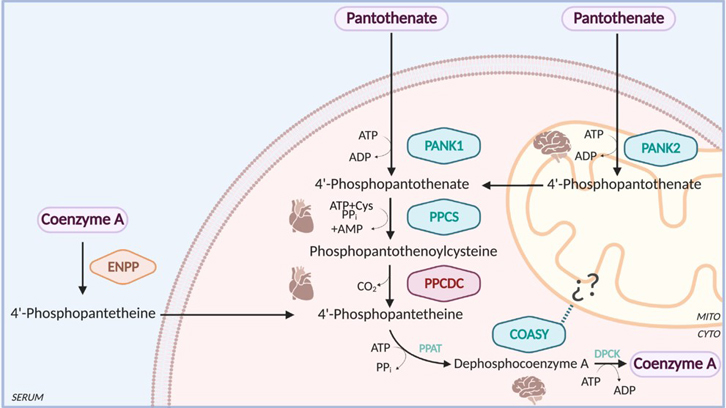
A study with the participation of researchers from the Yeast Molecular Biology group of the Institute of Biotechnology and Biomedicine (UAB) has described for the first time an ultra-rare congenital error in the metabolism of coenzyme A, involved in the processes of obtaining energy in the organism. This research lays the foundations for early detection of the error and its correction before it causes pathology.
Coenzyme A (CoA) is an essential cofactor involved in a wide variety of metabolic processes essential for obtaining energy. Both eukaryotes and prokaryotes synthesize CoA from pantothenate, also known as vitamin B5, through a nearly universal metabolic pathway. In humans, this pathway involves five enzymatic steps requiring four enzymes: pantothenate kinase (PANK), 4'-phosphopantothenoylcysteine synthetase (PPCS), phosphopantothenoylcysteine decarboxylase (PPCDC), and CoA synthase (COASY). To date, genetic defects were known in 3 of the 4 enzymes mentioned, the exception being PPCDC. In general, these defects gave rise to a severe pathology. PPCDC encodes a cysteine decarboxylase that uses flavin mononucleotide (FMN) as a cofactor and catalyzes the decarboxylation of 4'-phosphopantothenoylcysteine to 4'-phosphopantetheine. In humans, the native form is a 204 amino acid homotrimer in which the active site forms at the interface of every two subunits.
The first reported case of genetic defects associated with PPCDC was recently published in the Journal of Inherited Metabolic Disease as a result of a multicenter collaboration between various laboratories in Madrid and Valencia, and the participation of UAB researchers. This is a case detected in two sisters, who presented severe heart problems and died at a very young age, which could be associated with biallelic variants (p.Thr53Pro and p.Ala95Val) of PPCDC. These variants affect highly conserved residues across different species; p.Thr53Pro is involved in flavin mononucleotide binding, and p.Ala95Val is probably a destabilizing mutation. As a result, the fibroblasts derived from these patients showed an absence of PPCDC protein and an almost 50% reduction in CoA levels, and the cells presented clear problems of energy deficiency accompanied by defects in mitochondrial respiration.
These results were supported by the experiments carried out using a yeast model deficient in PPCDC and hence in CoA biosynthesis, developed by the Yeast Molecular Biology group of the Institute of Biotechnology and Biomedicine (UAB) under the direction of Dr. Joaquin Ariño. This is, therefore, a paper that describes for the first time a new and ultra-rare severe inborn error of metabolism caused by pathogenic variants of PPCDC. In this way, we lay the foundations for a possible early detection that would allow a supplementation therapy that would bypass the deficiency in PPCDC.
This work also highlights the importance of multidisciplinary collaborative studies in elucidating the genetic bases of ultra-rare diseases and emphasizes the importance of developing research models in unicellular eukaryotes, such as yeasts, whose application can be crucial in the investigation of more complex systems.
Department of Biochemistry and Molecular Biology, Universitat Autònoma de Barcelona.
Institut de Biotecnologia i Biomedicina (IBB-UAB), Universitat Autònoma de Barcelona.
References
Bravo-Alonso I, Morin M, Arribas-Carreira L, Álvarez M, Pedrón-Giner C, Soletto L, Santolaria C, Ramón-Maiques S, Ugarte M, Rodríguez-Pombo P, Ariño J, Moreno-Pelayo MÁ, Pérez B. Pathogenic variants of the coenzyme A biosynthesis-associated enzyme phosphopantothenoylcysteine decarboxylase cause autosomal-recessive dilated cardiomyopathy. J Inherit Metab Dis. 2022 Dec 23. doi: 10.1002/jimd.12584. Epub ahead of print. PMID: 36564894.